Sandra Waugh Ruggles, Juliana R. Perl, and Zachary Sexton, Stanford Byers Center for Biodesign; Kevin Schulman, School of Medicine, Graduate School of Business, Stanford University; and Josh Makower, Stanford Byers Center for Biodesign, School of Medicine, School of Engineering, Stanford University
Contact: jmakower@stanford.edu
Abstract
What is the message: The Medicare Coverage of Innovative Technology (MCIT), intended to provide four years of Medicare coverage for FDA-authorized breakthrough technologies, has become a source of controversy over procedural and evidentiary requirements. As a result, the Centers for Medicare and Medicaid Services (CMS) repealed the MCIT pathway for technological innovations. Yet a well-designed MCIT program that enables both coverage and continuing evidence collection could greatly accelerate patient access to important health advances and encourage invention and investment in areas of critically important unmet clinical needs.
What is the evidence: A survey of healthcare innovators and investors was designed and analyzed to better understand how MCIT, and the associated access to breakthrough technologies, might directly impact patients and the health technology innovation ecosystem.
Links: Supplemental Data
Timeline: Submitted: December 16, 2021; accepted after review: December 21, 2021.
Cite as: Sandra Waugh Ruggles, Juliana R. Perl, Zachary Sexton, Kevin Schulman, Josh Makower. 2022. The Need for Accelerated Medicare Coverage of Innovative Technologies: Impact on Patient Access and the Innovation Ecosystem, Health Management, Policy and Innovation (www.HMPI.org), Volume 7, Issue 1.
Introduction
The concepts behind the new Medicare coverage pathway, known as Medicare Coverage of Innovative Technology (MCIT), have been in development for almost 10 years and were published as a final rule as CMS-3372-F on January 14, 2021 with the intention of improving patient access to FDA designated breakthrough medical technologies.[1] However, on November 15, 2021, CMS issued a final rule repealing the MCIT program prior to its implementation.[2]
The original MCIT rule provides that any product participating in the Breakthrough Devices Program[3] at the Food and Drug Administration (FDA) would be granted immediate access to four years of Medicare coverage upon FDA authorization. A breakthrough designation is given to devices that meet specific criteria from the 21st Century Cures Act to “provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions” and fulfill at least one of these additional criteria:
- Represents a breakthrough technology
- No approved or cleared alternatives exist
- Offers significant advantages over existing approved or cleared alternatives
- Device availability is in the best interest of patients
Since Medicare beneficiaries are more likely to have life threatening or irreversibly debilitating diseases or conditions, they are more likely to benefit from access to breakthrough technologies that promise more effective treatment or diagnosis. As novel technologies, however, many breakthrough devices are unable to leverage existing reimbursement pathways, leaving patients waiting while sponsors engage in the time-consuming process of seeking new coding, coverage, and payment.[4],[5] The MCIT proposal was intended to streamline this process and incentivize companies to invent and commercialize breakthrough innovations that could have a large impact on patient outcomes and access.
While there is general consensus on the merits of an improved approach to the development and reimbursement of breakthrough technologies for Medicare beneficiaries, many aspects of the MCIT proposal were perceived as controversial. During the open comment period, concerns were raised around the strength of clinical evidence specific to the Medicare population, unmanageable costs, the difficulty of limiting or removing Medicare coverage for a device once it is established, how CMS might handle breakthroughs outside of existing Medicare benefit categories, and the definition of the current Medicare coverage requirement that a technology be “reasonable and necessary”.[6],[7],[8] Supporters of MCIT believe that many of these risks can be addressed with proper guardrails and further process/definitions added to the regulation.2 Rather than abandoning the pathway in response to concerns that have been raised, it is worth considering whether changes to the existing regulation could strengthen the approach while still providing more certainty around coverage, as well as incentives to bring more life-altering technologies to patients.
To help inform this discussion, we developed and distributed a survey to assess the need for a redesigned, streamlined reimbursement pathway such as MCIT for novel breakthrough devices. This survey focused on the existing reimbursement pathway for novel and breakthrough technologies and the time and development cost required after FDA authorization. The results also captured the perspectives of innovators and investors with experience in health technology development to help evaluate the impact MCIT would have on the innovation ecosystem and subsequent patient access to novel technologies. Further, we selected a small number of exemplar technologies that FDA has designated as breakthrough devices to show how the acceleration of patient access might impact individual health outcomes and overall healthcare system costs once such devices demonstrate safety and efficacy sufficient for FDA authorization.
Study Data and Methods
Study Design
Our study assessed current and future perceptions of reimbursement timelines using a voluntary survey of healthcare innovators with expertise in reimbursement processes for breakthrough technologies. Respondents were recruited through invitations emailed to the Stanford Byers Center for Biodesign communication list, as well as to members of the Advanced Medical Technology Association (AdvaMed), the Medical Device Manufacturers Association (MDMA), the National Venture Capital Association (NVCA), and several additional health technology associations including BioUT, LifesciencesPA, MASSMedic, and WASHBio. The survey was designed and hosted using the Qualtrics XM platform[*] and was available from September 26, 2021 through October 7, 2021. Two complementary sets of survey questions were used: one targeted at innovators (individual inventors, experts, and knowledgeable employees of large and small entities engaged in bringing innovative technologies to market) and one targeted at investors who fund the development and commercialization of innovative medical technologies.
Sample. 497 respondents opened the survey through an anonymous link and 381 respondents completed the survey. Partial responses were not included in the data analysis to preserve a consistent cohort of respondents. Unique IP addresses were collected to ensure one response per person. Respondents were screened for experience with the reimbursement process and knowledge of the timing for achieving coverage of new medical technologies by eliminating respondents who self-selected an expertise level below 3 (from a range of 0 to 10). The sample was further restricted to respondents who self-selected a professional role as a healthcare investor, innovator, or industry reimbursement expert. In total, 253 innovators and 83 investors were included in our analysis.
Data Analysis. Results were viewed using the report feature in the Qualtrics survey tool and analyzed using Microsoft Excel. In addition, sub-group analysis was used to detect whether statistically significant differences in responses existed for specific sub-groups, including investors who primarily focus on medical device and diagnostics investments, innovators at start-up companies, industry experts at mid- or large-sized companies, and respondents who self-identify as reimbursement or market access professionals.
Exemplar Technologies and Economic Impact Calculations
We chose a small sample of breakthrough technologies to illustrate the potential patient impact of products in development. We selected these technologies from a list of companies that had publicly announced that their products had received breakthrough product designation by FDA within the previous five years. We refined our choices to include technologies that primarily impact Medicare patients and are in clinical categories where Medicare spending is significant. Each company was contacted to learn more about their technology. We also tracked all publicly disclosed product development milestones. The patient impact of each technology was estimated independently using published sources. Where multiple sources were available, or variability existed in published data, a range is provided. Specific calculations are detailed in the appendix.
Limitations
The study design has the following notable limitations. First, the respondent pool for the survey reflects the characteristics of the organizations who promoted it and primarily includes industry leaders who are focused on the development and commercialization of innovative healthcare products, as well as investors who have made healthcare investments despite the risks and complications inherent in the field. Second, the respondents represent a highly motivated cross-section of the health technology landscape. 87% of responding investors had at least one investment in a company developing a product with (or pursuing) breakthrough designation, and 76% of industry respondents are currently working on one or more products with (or pursuing) breakthrough designation. While these respondents are knowledgeable of the MCIT proposal and the current reimbursement environment, they also are those most likely to benefit from the streamlined process proposed in the regulation.
Study Results
Characteristics of Respondents
Innovators and industry experts. The innovators in the sample (Supplemental Data) represent those most heavily engaged in the development of breakthrough medical products. Respondents had an average of 22 years of experience in healthcare and have worked in an average of 3.2 clinical areas in their careers, with the most common area being cardiovascular disease (65%). Experience in oncology, neurological disease, neurovascular disease, pulmonary disease, and endocrine disease was also common in the sample. The majority of respondents were from start-up companies (50%) with fewer than 50 employees. 84% of the sample were in executive leadership roles or in reimbursement or market access roles.
Healthcare investors. Demographic information also was collected from 83 healthcare investors (Supplemental Data). In aggregate, the responding investors were managing an average of $1.4 billion in investment capital. 55% managed between $100 million and $500 million in investment capital, with two investors holding $40 billion or more in funds dedicated to healthcare. 41% of investors indicated that a significant percentage of their investments (more than 50%) were devoted to medical devices or diagnostics products, including 7% who invest exclusively in diagnostic devices. Similar to the innovators in the sample, investors who completed the survey tended to have already invested in companies pursuing breakthrough product designation with the FDA. Fewer than 10% of investors had not yet made an investment in a company pursuing a breakthrough product, while over half (61%) of investors had three or more investments in such companies.
Perceptions Of The Current Reimbursement Environment
Timelines. Innovators were asked about the typical timeline to specific reimbursement milestones for novel and breakthrough products following FDA authorization (Figure 1).
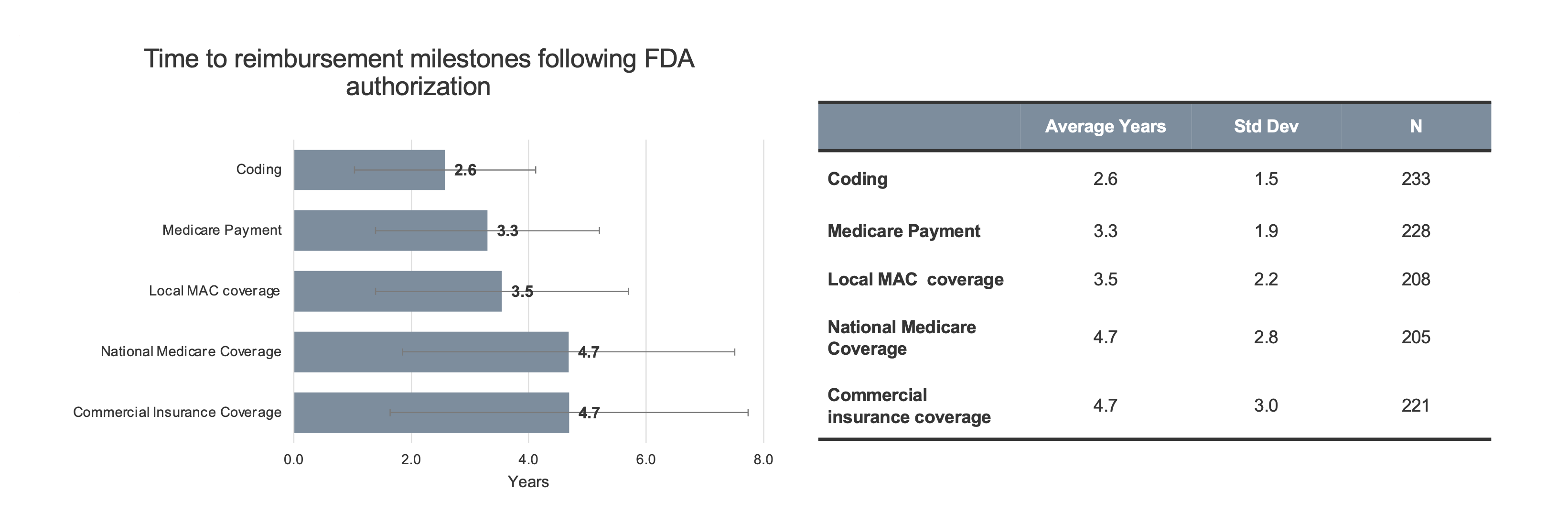
Figure 1. Innovator estimates of time to reimbursement milestone. Respondents were asked to estimate, in their experience, the time to each reimbursement milestone. Respondents were allowed to leave entries blank if they did not feel comfortable providing an estimate based on their experience. Therefore, N varies. Coding: N=233 (92%), Medicare payment: N = 228 (90%), Local MAC coverage: N=208 (82%), National Medicare coverage: N=205 (81%), and Commercial Insurance Coverage: N=221 (87%) of respondents answered. Data from one respondent was excluded for inputting 0 for all fields, data from two additional respondents was excluded for inputting >50 years for Coding.
Respondents were allowed to leave timeline options blank, as needed, to reflect their experience. However, data was collected from over 80% of the respondents for each timepoint. As shown in the figure, the average time to acquire a new code was reported to be 2.6 years (std dev 1.5 years). The time to establish Medicare payment was 3.3 years (std dev 1.9 years). For a local Medicare Administrative Contractor (MAC) coverage policy, also referred to as a local coverage determination (LCD), the average time was 3.6 years (std dev 2.2 years). To achieve nationwide Medicare coverage, the average time was 4.7 years (std dev 2.8 years).[†] That was virtually equivalent to the time required for widespread commercial insurer coverage (4.7 years, std dev 3.0 years). Sub-group analysis resulted in markedly different timelines for only one sub-group of respondents – those whose experience was diagnostics reimbursement (n=17). These respondents indicated shorter timelines to coding (1.3 years versus 2.6 years), Medicare payment (2.2 years versus 3.3 years), local MAC coverage (2.3 years versus 3.5 years), and national Medicare coverage (2.4 years versus 4.7 years) compared to the entire innovator cohort.
Write-in comments. To better understand each innovator’s personal experience with reimbursement timelines, open-ended comments were collected in the survey. 153 respondents (61%) provided qualitative responses. Their feedback exemplifies the concern and frustration felt among innovators about current reimbursement processes.
Real-world evidence collection. To assess the impact that requiring further clinical data or the collection of real-world evidence on Medicare patients after FDA authorization might have on innovators utilizing an MCIT-like program, innovators were asked about the frequency with which their companies normally collect clinical data after FDA authorization (Figure 2).
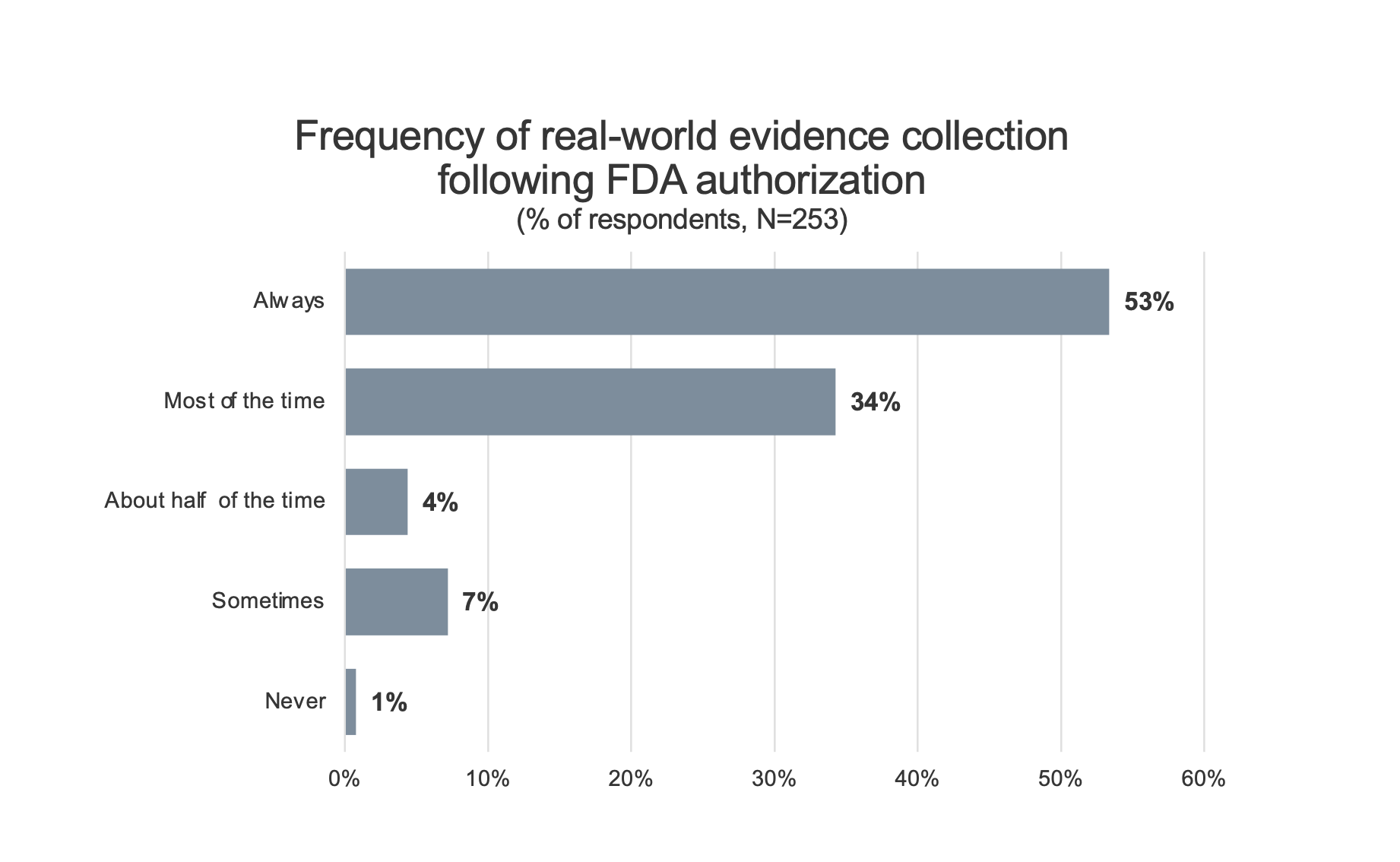
Figure 2. Innovator reported frequency of real-world evidence collection after FDA authorization for a novel or breakthrough technology. Respondents indicate that it is frequent for companies to collect additional clinical evidence following FDA authorization. 53% of respondents indicate that their firm “always” collects such data.
87% of respondents indicated that collecting additional clinical data or real-world evidence was something that they do (53% always, 34% most of the time) as part of the commercialization of a breakthrough product. Only 1% of respondents indicated that they do not routinely collect some form of real-world evidence following FDA authorization for a novel technology.
Investment risks. To ascertain the relative weight of the risks that investors consider when deciding whether to back breakthrough technologies, responding investors were asked to rank a list of issues that might affect their decision to invest in a particular company (Supplemental Data). Three external risk factors (reimbursement pathway, regulatory pathway, and availability of capital) dominated the concerns of investors, with 55% of investors ranking the reimbursement pathway as the greatest external risk factor followed by the regulatory pathway (30%).
Effectiveness of current accelerated pathways. To understand the degree to which existing programs like the Program for Parallel Review of Medical Devices or the Coverage with Evidence Development (CED) program address innovators’ needs for accelerated coverage, respondents were asked to rate the sufficiency of these initiatives (Supplemental Data). Respondents in large part felt that the current programs were not sufficient to support breakthrough product designation, with 54% of innovators and 79% of investors disagreeing with the statement: “The existing parallel review process with FDA and the CED pathway are sufficient to provide timely patient access for novel medical technologies.” Reasons for this are cited in the discussion section.
Likelihood of pursuing breakthrough product innovation. To determine the extent to which an MCIT-like program would stimulate the advancement of breakthrough technologies, innovators were asked how likely they would be to take on such a project if an accelerated program was in place.
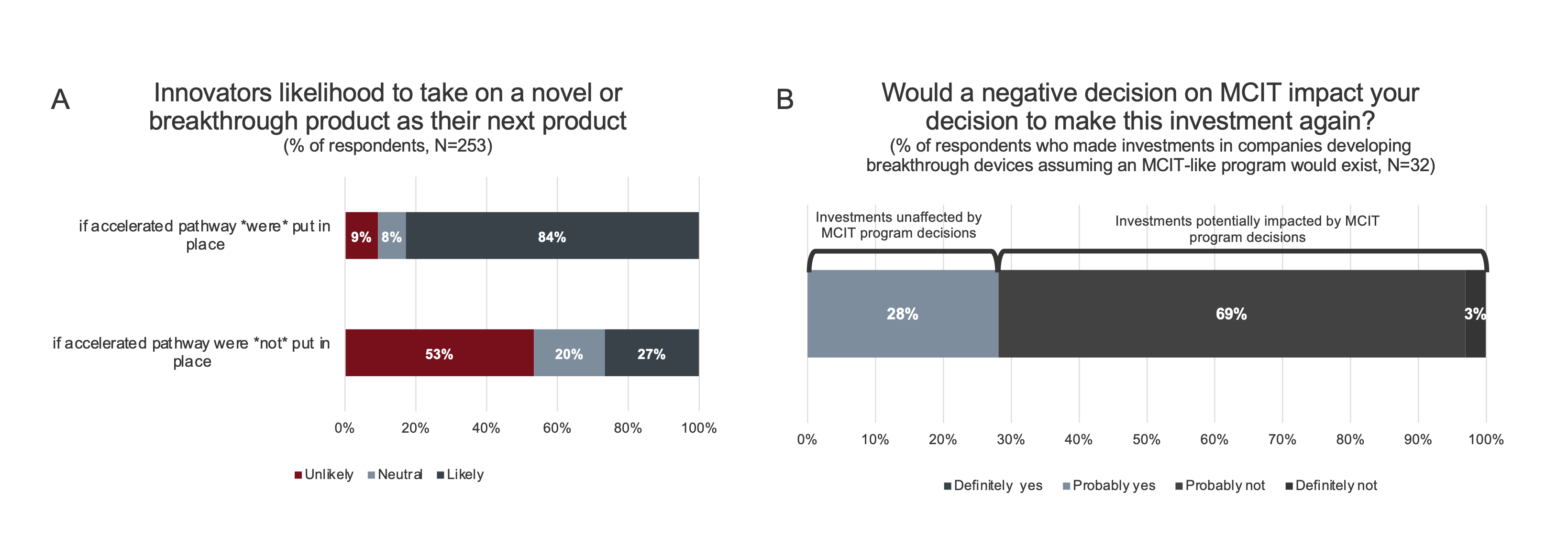 Figure 3. Likelihood to undertake novel or breakthrough product innovation. A. Innovators are incentivized to undertake high-risk novel and breakthrough product innovation with the introduction of an accelerated pathway for Medicare coverage. Responses for “Highly likely” and “somewhat likely” were consolidated, as were responses for “highly unlikely” and “somewhat unlikely”. B. Investors were asked if their previous investments in companies developing novel and breakthrough technologies were made on the assumption that an accelerated pathway for CMS coverage would exist. 46 respondents responded No. Of the 32 respondents who responded yes, a subsequent question was asked: “On the whole, would you make these investments again knowing that a new program (like MCIT) that expedites Medicare patient’s access would not be available?”
Figure 3. Likelihood to undertake novel or breakthrough product innovation. A. Innovators are incentivized to undertake high-risk novel and breakthrough product innovation with the introduction of an accelerated pathway for Medicare coverage. Responses for “Highly likely” and “somewhat likely” were consolidated, as were responses for “highly unlikely” and “somewhat unlikely”. B. Investors were asked if their previous investments in companies developing novel and breakthrough technologies were made on the assumption that an accelerated pathway for CMS coverage would exist. 46 respondents responded No. Of the 32 respondents who responded yes, a subsequent question was asked: “On the whole, would you make these investments again knowing that a new program (like MCIT) that expedites Medicare patient’s access would not be available?”
As shown in Figure 3A, 87% of innovators reported they would be more likely to take on a breakthrough technology as their next project if an MCIT-like accelerated program was in place versus 27% without such a program. Understanding that some clinical fields are more challenging for developing novel technologies than others, the survey further asked innovators in which fields would they be more likely to advance innovations if an MCIT-like program was available. While all of the noted clinical fields would be helped by such a program, the largest positive impact was on cardiovascular diseases (52%), which is the field most likely to have a majority of Medicare beneficiaries (Supplemental Data).[9]
Likelihood of investing in breakthrough product innovation. To assess how investors would react to an MCIT-like program, the subset of investors who have invested in companies with breakthrough products (94% of respondents, N=78) were questioned about their past investment decisions (Figure 3B). While the majority of these investors (59%, N=46) did not base their investment decisions on the existence of an MCIT-like program, a subset (29%, N=32) reported that they would not have made those investments without the promise of such a program. In addition, all investors were asked how likely they would be to increase their amount of investment in breakthrough products if an MCIT-like program were to be available. 84% of the investors surveyed reported that they would increase their investments, with the largest positive impacts on potential investments in neurovascular disease/stroke, neurological disease, and cardiovascular disease (Supplemental Data).
Patient and Healthcare System Impact
Breakthrough Example 1: Reducing bleeding complications in cardiothoracic surgery
Patients with heart disease are typically prescribed combinations of antithrombotic medicines, including anti-platelet drugs such as ticagrelor, to reduce the risk of dangerous blood clots that can lead to heart attacks, strokes, or even death. These same patients often need cardiac surgery. However, anti-platelet drugs substantially increase the risk of major bleeding events, and standard clinical practice advises postponing surgeries until at least 72 hours after the last dose to allow anti-platelet drugs to be washed out of the body.[10] Yet approximately 50,000 patients in the United States on ticagrelor undergo emergency cardiac surgeries annually and cannot wait through this washout period, putting them at uniquely higher risk.[11],[12] As many as 65% of these patients experience serious or life-threatening bleeding events, with a 4.5x higher mortality rate.9,[13],[14] The added economic burden of bleeding events ranges between $7,000 and $29,000 per patient, depending on the surgical complexity of the procedure.[15],[16],[17]
In April 2020, the FDA granted breakthrough designation to CytoSorbents’ DrugSorb-ATR system, a single-use disposable device to remove the antiplatelet drug ticagrelor from patients during emergency open heart surgeries.[18] In an ongoing 120-patient randomized, controlled FDA pivotal trial (STAR-T; NCT04976530), patients undergoing on-pump cardiothoracic surgery less than 48 hours after the last dose of ticagrelor were evaluated to assess the DrugSorb-ATR system’s ability to reduce perioperative bleeding events and blood ticagrelor levels.[19] The study size and expected outcomes were based on real-world data outcomes in the European Union, where this device was approved for on-pump cardiothoracic surgeries in January 2020.[20] Based on the study results, the device is expected to reduce circulating ticagrelor levels by at least 40% and confer clinically meaningful reductions in moderate, severe, and massive bleeding.15,[21] Accordingly, it is estimated that more than 13,000 major bleeding events, primarily among Medicare beneficiaries, could be eliminated each year if pivotal trials validate the earlier findings (see Supplemental Data for calculation). In the future, the targeted beneficiary population will likely increase because the DrugSorb-ATR system received a second Breakthrough Device Designation in August 2021 for the direct oral anticoagulant (DOAC) drugs apixaban and rivaroxaban when used during urgent cardiac surgery.[22] A second pivotal trial to collect evidence of bleeding outcomes in target subjects treated with indicated DOACs is anticipated to begin enrollment in early 2022 (STAR-D; NCT05093504).[23]
Breakthrough Example 2: Early diagnosis of skin cancer
Skin cancer is the most common cancer in the United States, impacting an estimated one in five Americans in their lifetime.[24],[25],[26] In 2021, almost 200,000 new cases of the deadliest skin cancer, melanoma, will be diagnosed.[27],[28] The prognosis for melanoma patients diagnosed early is promising (5-year survival rate of 99%).[29] However, once the cancer spreads, survival drops dramatically (5-year survival rate of 27%). 5.2 million biopsies were performed in the Medicare population in 2016 (from 1.4 million in 1993) and a 2.1:1 ratio for skin biopsy-to-skin cancer treatment (up from 1.1:1 in 1993) indicates that the threshold for ruling out of melanoma is low.[30]
Veriskin’s TruScore is a non-invasive handheld device with FDA Breakthrough Device Designation for real-time diagnosis of skin cancers. The technology assesses skin vascular networks to determine the likelihood that a lesion is cancerous. A pilot study demonstrated sensitivity of over 99% and specificity of 94% in a sample of 125 biopsy-verified lesions.[31] This technology has the potential to significantly decrease the number of biopsies performed for non-cancerous, suspect lesions while increasing the accessibility of cancer screening by enabling it to be performed by non-specialists. More than 92% of adults over 65 years old have an annual wellness visit.[32] Yet data shows that only about 8% of patients who had seen a primary care physician (PCP) or an obstetrician/gynecologist in the past 12 months had received a skin examination, for which the gold standard is a subjective visual examination.[33] Thus, if validated by the upcoming pivotal studies, implementation of the Veriskin TruScore technology has the potential to save lives by first facilitating earlier skin cancer diagnosis and secondarily by reducing referrals to dermatologists for non-cancerous lesions.
Breakthrough Example 3: Treatment of hypertension
Three out of four seniors in the United States have hypertension.[34] This epidemic was the primary underlying cause of >190,000 deaths in 2019.[35] Economically, hypertension is estimated to cost the US healthcare system $50 billion each year for medication, physician visits, and hospitalizations.[36],[37] Resistant hypertension, or treatment-resistant hypertension, occurs in 16% of US patients who are consequently at high-risk for adverse cardiovascular events.[38],[39]
ReCor Medical’s Paradise System is an FDA-designated breakthrough device that delivers high-intensity, focused ultrasound energy via a catheter to destroy nerves along the renal artery. Called renal denervation, this new, minimally-invasive procedure treats hypertension by decreasing renal sympathetic nervous system activity. The company has completed three clinical studies to date, including the 282-person RADIANCE-HTN study. This study demonstrated a >5 mmHg decrease in daytime systolic blood pressure in two out of three patients treated.[40],[41],[42] Responders with resistant and non-resistant hypertension experienced average blood pressure decreases of 17 mmHg and 14 mmHg, respectively. For comparison, a large meta-analysis of medication effectiveness demonstrated that a 10 mmHg reduction in systolic blood pressure reduced the risk of major cardiovascular events by 20%, and all-cause mortality by 13%.[43] Thus, if further real-world data agrees with previous clinical studies and renal denervation using the ReCor Medical Paradise System reduces blood pressure by at least 10 mmHg, patients treated would experience fewer major cardiovascular events and enjoy longer lives – with up to 16,000 lives saved annually upon integration into the routine care of these patients (see Supplemental Data for calculation).
Breakthrough example 4: Early diagnosis of pancreatic cancer
More than 60,000 cases of pancreatic cancer will be diagnosed in the US in 2021, with fewer than 6,000 patients (11%) surviving beyond five years.[44],[45],24 As with all cancers, early pancreatic cancer detection greatly improves the odds of successful treatment. However, in 80% of pancreatic cancer diagnoses, the cancer has already spread to surrounding organs.40 Ensuing attempts to treat Medicare beneficiaries, specifically, with metastatic pancreatic cancer end up costing approximately $40,000 per patient.[46],[47]
In March 2021, the FDA granted a Breakthrough Device Designation to a novel liquid-biopsy pancreatic cancer diagnostic device from Bluestar Genomics. The company’s goal is to provide an early-stage pancreatic cancer screening option for newly diagnosed diabetic patients,[48] That would potentially be relevant to more than one million patients annually. This is acutely important as ~25% of pancreatic cancer diagnoses are preceded by the development of diabetes.[49] Within the Medicare-age population, there is an estimated diabetes incidence of about 326,000 new cases per year.[50] To inform the predictive capacity of Bluestar’s technology, a retrospective case-control study was conducted to determine different epigenomic signatures that are characteristic of pancreatic cancer. The results showed that the enrichment and absence of 5-hydroxymethyl-cytosine (5hmC) can accurately inform predictive diagnostic models for pancreatic cancer.[51],[52] In subsequent feasibility studies, 176 tissues from breast, colorectal, lung, ovary, and pancreas (44 per tumor type and 11 per stage I-IV along with 10 healthy tissues per type) were used to evaluate the performance of this technology for cancer detection. From this study, the cross-validation sensitivity, an important performance metric of predictive models, was found to be 56% at 99% specificity for pancreatic cancer, providing further evidence of strong diagnostic capabilities for Bluestar’s technology.[53] In combination, this early work demonstrates the potential to identify 5,000-10,000 pancreatic cancer cases annually in this high-risk population (see Supplemental Data for calculation), filling a much-needed clinical gap where the only current alternatives are image-based tests that perform poorly at early stages of the disease.
Discussion
Important Takeaways
The innovators and investors who responded to our survey were knowledgeable of United States reimbursement processes and familiar with breakthrough products. Interestingly, the estimates for reimbursement timelines exhibited more variability than might have been expected given this expertise (Figure 1). For example, length of time to establish coding was 2.6 +/- 1.5 years and the length of time to local MAC coverage was 3.5 +/- 2.2 years. Innovators with expertise in diagnostics reimbursement indicated that the reimbursement timeline was up to 1 year shorter for diagnostic products. For important breakthrough therapeutic technologies, however, it takes 4.7 +/- 2.8 years to establish nationwide coverage. This is a devastatingly long time for patients, as well as for small, venture-backed companies without deep financial resources to sustain their operations through this period. For investors, the majority of investor respondents consider the reimbursement pathway to be the highest-impact external risk factor to an investment. Consequently, the uncertainty of the timelines to achieve coding, coverage, and payment create a much higher bar for investment in important clinical areas and a strong disincentive for investment in breakthrough products. As a result, these delays, and the lack of potential payment for breakthrough technologies, have a direct impact on patients who must go without leading-edge interventions.
The open-ended comments captured the reasons respondents felt they experienced such variety in timelines to reimbursement. Innovators describe a reimbursement environment that is “challenging,” “circular,” and “highly variable.” As one respondent described, “Coding requires wide utilization. Wide utilization requires coverage and payment. Payment requires coding, particularly physician payment. And without coverage, [it is] difficult to get 10% of a prevalence pool treated to meet the wide utilization criteria. [The] system is designed to halt any innovation.” A responding investor stated, “Without clear guidance and agreements early on, the system penalizes innovation and rewards incremental changes. It is hard to fund start-ups with breakthrough technologies because of the reimbursement challenges.”
One example of a common reimbursement challenge highlighted in the comments is the transition from temporary Current Procedural Technology (CPT®) codes to established CPT codes. Temporary CPT codes (also called Category III codes) are granted by the American Medical Association (AMA) for emerging technologies, services, or procedures.[54] These codes are not assigned a payment amount, and technologies that use these codes are frequently categorized as “experimental and investigational” by commercial insurers with associated non-coverage decisions. Innovators describe a payment landscape where the only mechanism for reimbursement is case-by-case adjudication from a limited group of payers in a highly resource-intensive fashion.[‡] Thus, the company enters a “valley of death” where they are simultaneously expected to build evidence of widespread product utilization and real-world clinical evidence to support permanent codes in a landscape where the majority of insurers do not provide payment for the temporary code, and those that do, require resource-intensive appeals by providers and their patients. Innovators described timelines to achieve Category I codes that stretched from three years to more than a decade.
Additionally, products with new CPT codes also require either a positive LCD issued by local MACs or a national coverage determination (NCD) from CMS to provide hospitals and physicians with ongoing payment. These decisions are governed by a process that includes statutory requirements for the timely review and input from patients, providers, and the developer of the technology.[55] However, the average time to a local MAC coverage decision reported by responding innovators was 3.5 +/- 2.2 years. One innovator’s comment highlighted this apparent disconnect as follows, “In our case, commercial coverage came MUCH more rapidly than Medicare coverage. Securing Medicare coverage took more than a decade of advocacy.” Another innovator observed, “The MACs are overburdened, and the local coverage process now takes almost as long as the NCD process. CMS’s Coverage and Analysis Group is in desperate need of additional resources and only can conduct 4-6 national coverage assessments a year. New technologies can wait in the coverage queue for years.” Thus, the statutory requirements for a timely LCD or NCD may be met once a technology is assessed, but the timeline to a successful, positive coverage decision for a breakthrough product may take substantially longer.
Issues with Current Programs
Innovators and investors alike do not feel that current programs to accelerate coverage such as parallel review and CED are sufficient to support timely access to technology (Supplemental Data). The original intent of Medicare’s CED pathway was to accelerate patient access, specifically acknowledging this need. Unfortunately, CED requires manufactures to enter into the NCD pathway to participate, forcing innovators to make an existential decision. Specifically, while a positive NCD opens up the US market via CED and nationwide coverage, a negative NCD (or the decision to reject an NCD and revert decisions to the local MACs) can result in a “walking-dead” state for a new technology. A negative NCD can create a perception that the technology is still “experimental” and, as such, both local MACs and private insurers may deny coverage for many years, if not indefinitely. It is important to emphasize that the limited impact of the current CED pathway arises from the absence of a clear statutory direction and not from inherent conceptual flaws. In practice, CMS decision memos reflect difficulties in applying evidence-based coverage authority when evaluating CED devices.57 Often CMS interprets “reasonable and necessary” as “adequate evidence to conclude that the item or service improves health outcomes,” but this directly conflicts with the goal of evidence-collection during CED, placing novel technologies in a catch-22 for NCDs and other coverage decisions.[56] A more effective CED program, such as MCIT, needs to be based on the potential for new technologies to be “reasonable and necessary” since it is in Medicare’s interest to cover such technologies while they develop more comprehensive evidence.
Addressing MCIT Concerns
While the potential benefits of an accelerated pathway for breakthrough technologies that delivers improved patient outcomes and access are clear, the issues raised by opponents of MCIT must be addressed. First, in the previous MCIT proposal, there was no mandate for post-market studies to ensure that real-world evidence will continue to support Medicare’s early coverage decision once the four-year period runs out. Further, since some new technologies or procedures offer benefits to patients outside of the Medicare population, clinical studies may not include enough patients in the Medicare population to render a full conclusion with respect to how widely such a technology may be applied. In response to these concerns, it would be reasonable to modify MCIT to include a requirement for post-market clinical studies and/or real-world evidence to be collected specifically in the Medicare population. Since 87% of the innovators and industry experts surveyed reported that they routinely collect such data post-market, adding such a requirement would not represent an undue burden.
Another issue was that the previous MCIT program could have made it harder for CMS to withdraw coverage if the device or procedure later turned out to be unsafe or ineffective. Withdrawing coverage can be difficult because of pressure from the company, healthcare providers, and patients. However, the annual rule-making process is just one example where CMS has experience carefully considering the evidence, taking input from stakeholders, and making difficult decisions. A revised MCIT program could include a mechanism whereby coverage can be withdrawn or limited due to a safety issue through a well-designed process and clear decision-specific metrics or criteria. CMS is well-positioned to make appropriate decisions if a technology or procedure turns out to be unsafe or not cost-effective for Medicare beneficiaries.
One further concern was that the potential number of breakthrough technologies designated by the FDA could overwhelm CMS and be cost-prohibitive to Medicare. Conversely, breakthrough technologies typically offer high value to the healthcare system by improving the patient health outcomes-to-cost ratio. CMS has demonstrated their interest in incentivizing value-focused care through payment models introduced via the CMS Innovation Center (CMMI). Over the course of a Medicare beneficiary’s lifetime, major cost savings can be realized through the prevention of surgical complications, early-stage disease treatments, and other breakthrough solutions to irreversible problems. For example, if the ReCor technology successfully decreases cardiovascular events by 20%, then Medicare no longer has to pay for the expensive care necessitated by such events. Similarly, thousands of Medicare patients would realize better outcomes while the healthcare system incurs lowers costs through the use of technologies such as CytoSorbents’ DrugSorb-ATR system, Veriskin TruScore, or Bluestar Genomics’ diagnostic. The 4.7 +/- 2.8 year delay in achieving nationwide Medicare coverage takes on even deeper meaning when those delays are multiplied by the yearly death rates from those diseases. These four technologies alone have the potential to save hundreds of thousands of lives. Taken in combination, the possibility of decreased long-term spending and improved health outcomes creates a tremendous value proposition.
A final concern raised by critics of MCIT was that a breakthrough device might not have been tested in the Medicare population during development, and might have a different risk/benefit profile for the Medicare population. CMS has raised this concern when making previous coverage decisions for new medical devices[57], and this is an important challenge for proponents of the MCIT rule. In many cases, populations recruited to a clinical trial for the assessment of a treatment benefit might not be an ideal population for the assessment of generalizability to a population like the Medicare population. However, from a clinical epidemiology perspective, this is a core challenge in any type of clinical research. Few trials are designed solely to assess treatment benefit, so efforts to increase the generalizability of the dataset can be fraught with risk. In this case, does generalizability mean including Medicare patients, demonstrating the same treatment effect in the Medicare patient subgroup (often an underpowered assessment from a statistical perspective), or demonstrating a treatment effect specifically within the Medicare population by essentially requiring a new clinical trial of the same or larger size than the original? Further, these issues around Medicare patients extend to issues of race, gender, and treatment patterns, as well as issues of ethics (are Medicare patients interested in participating in an investigational clinical trial?) and futility (can the required subjects be recruited to the study in a reasonable time period?). Despite these limitations, it seems reasonable to suggest that MCIT be amended to include real-world evidence collection in at least a subset of the Medicare population post-approval.
Unfortunately, there were other procedural elements missing in the original MCIT proposal, which made it difficult for CMS to successfully adopt. Mark McClellan and colleagues at the Duke-Margolis Center in their November 1, 2020 comments to CMS[58] outlined a comprehensive set of modifications that we believe would substantially address a majority of the issues raised and provide a more rigorous framework that allows MCIT to achieve substantial value for Medicare patients. The Duke-Margolis Center proposal thoroughly addresses an approach towards follow-on devices, new codes and pass-through payments, guidance on evidence collection, achieving an effective transition from MCIT to long-term coverage after four years, operating procedures for CMS, and a call for more resources for CMS to effectively implement and sustain the pathway. The only major area where we substantially differ in our recommendation relates to the timing of when the certainty of being included in the MCIT program would be confirmed by CMS. In order for MCIT to act as a stimulus for breakthrough innovation and investment, the process must provide early certainty that the technology is included in the MCIT program. This should be determined at or near the time that breakthrough status is granted, which is usually before the pivotal trials have begun. Even if an additional step is needed after breakthrough determination, where CMS and the manufacturer work together to align on requirements for pre or post-market evidence development, it is essential for companies and investors to have certainty about post-authorization coverage. This early certainty would inform and motivate their investment in the costly pivotal and post-market studies that lie ahead. Proposals that delay this certainty or create new uncertainties that might play out after pivotal study completion or FDA authorization do not offer the innovation ecosystem the same powerful stimulus.
Conclusion
Breakthrough technologies, by definition, have the potential to bring better outcomes to patients suffering from life-threatening or irreversibly debilitating diseases or conditions, and thus have the potential to achieve healthcare’s triple aim of better care, better health, and lower cost. According to the data we have gathered, it takes breakthrough medical technologies an average of 4.7 years to achieve adequate coding, coverage, and payment by Medicare after proving safety and efficacy and receiving market authorization from the FDA. For many patients, this delay in receiving therapies that promise substantial life-saving or morbidity-reducing benefits is frustratingly too long. Since breakthrough technologies are often more challenging to bring through development, clinical studies, and reimbursement, investors and innovators alike are less likely to pursue such projects, impacting important fields such as cardiovascular disease, stroke, and cancer. The need to accelerate patient access to breakthrough products through an MCIT-like program is clear. We urge innovators, investors, and policymakers at CMS to work together to refine the MCIT proposal to help stimulate the creation and advancement of breakthrough technologies and prevent Medicare patients from having to wait years for these important therapies.
Disclosures
No authors received research funding for this work. SWR declares compensation for product design and business strategy consulting services – none for clients with products designated breakthrough medical devices. KS, JRP and ZAS have nothing to disclose. JM declares a compensated advisory and consulting relationship with a diversified investment firm that holds some investments in companies that have received breakthrough status. As part of that relationship, he also serves on the board of two companies with products that have received breakthrough status. None of those companies were highlighted in this work.
Acknowledgements
The authors wish to thank Paul Yock, Emeritus Director, Lyn Denend, Director for Academic Programs, Stacey Paris McCutcheon, Manager of Academic Projects and Communications, and Cece Torres, Program and Graphic Design Coordinator at the Stanford Byers Center for Biodesign for their help editing the article. We also wish to thank the leadership and members of MDMA, NVCA and AdvaMed for their help in distributing and participating in the survey and providing commentary and feedback. Thanks also to Dan Waldmann, Parashar Patel, Deneen Vojta, Larry Leisure, Justin Klein, and Ross Jaffe for their valuable input. Lastly, we wish to acknowledge the leadership of Cytosorbents, Inc., Bluestar Genomics, Inc., ReCor Medical, Inc. and Veriskin, Inc. for their help obtaining critical information about their technologies to support the article.
Notes
[*] The survey design, fielding and data collection for this paper was generated using Qualtrics software, Version October 2021 of Qualtrics. Copyright © 2021 Qualtrics. Qualtrics and all other Qualtrics product or service names are registered trademarks or trademarks of Qualtrics, Provo, UT, USA, https://www.qualtrics.com.
[†] The survey used the phrase “national Medicare coverage” to encompass a National Coverage Determination (NCD) and nationwide coverage through the accumulation of local MAC coverage decisions.
[‡] Traditionally, Medicare does not allow payment by prior authorization, so a series of local coverage determinations (LCD) or a national coverage determination (NCD) is required for payment. More recently, Medicare Advantage plans have implemented prior authorization policies.
References
[1] Medicare Coverage of Innovative Technology (CMS-3372-F) Fact Sheet. Centers for Medicare and Medicaid Services, January 2021. https://www.cms.gov/newsroom/fact-sheets/medicare-coverage-innovative-technology-cms-3372-f
[2] Medicare Program; Medicare Coverage of Innovative Technology (MCIT) and Definition of “Reasonable and Necessary.” 86 FR 51326, September 15, 2021. https://www.federalregister.gov/documents/2021/09/15/2021-20016/medicare-program-medicare-coverage-of-innovative-technology-mcit-and-definition-of-reasonable-and
[3] Breakthrough Devices Program. US Food & Drug Administration. Updated January 5, 2021. Accessed October 14, 2021. https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program#s8
[4] Fudim M, Abraham WT, Bardeleben RS, et al. Device therapy in chronic heart failure: JACC State-of-the-art review. JACC. 2021 Aug;78(9):931 Published 2021 Aug 31. doi: https://doi.org/10.1016/j.jacc.2021.06.040
[5] Knight BP, Deering TF, Gold MR, Mittal S. The problematic lag between FDA approval of medical devices and CMS coverage. J. Cardiovascular Electrophysiology. 2021 May;32(7):1801. Published 2021 May 30. doi: https://doi-org.stanford.idm.oclc.org/10.1111/jce.15112
[6] Medicare Program; Medicare Coverage of Innovative Technology (MCIT) and Definition of “Reasonable and Necessary.” 86 FR 51326, May 18, 2021. https://www.federalregister.gov/documents/2021/05/18/2021-10466/medicare-program-medicare-coverage-of-innovative-technology-mcit-and-definition-of-reasonable-and – print
[7] Neumann PJ, Chambers JD. Eroding Progress On Evidence And Outcomes: CMS’s New Proposed Pathway For Medical Device Coverage. Health Affairs Blog, December 2020. https://www.healthaffairs.org/do/10.1377/hblog20201130.767638/full/. 10.1377/hblog20201130.767638
[8] Bach PB. After 4 Years of Trump, Medicare and Medicaid Badly Need Attention. New York Times, December 2020. https://www.nytimes.com/2020/12/01/opinion/trump-medicare-medicaid.html
[9] Rodgers JL, Jones J, Bolleddu SI, et al. Cardiovascular Risks Associated with Gender and Aging. J Cardiovasc Dev Dis. 2019;6(2):19. Published 2019 Apr 27. doi:10.3390/jcdd6020019
[10] Hillis LD, Smith PK, Anderson JL, et al. 2011 ACCF/AHA guideline for coronary artery bypass graft surgery: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines [published correction appears in J Thorac Cardiovasc Surg. 2012 May;143(5):1235]. J Thorac Cardiovasc Surg. 2012;143(1):4-34. doi:10.1016/j.jtcvs.2011.10.015
[11] Mitrev Z, Anguseva T. Emergencies in cardiovascular surgery. J Cardiothorac Surg. 2013;8(Suppl 1):O2. Published 2013 Sep 11. doi:10.1186/1749-8090-8-S1-O2
[12] Alkhouli M, Alqahtani F, Kalra A, et al. Trends in Characteristics and Outcomes of Patients Undergoing Coronary Revascularization in the United States, 2003-2016 [published correction appears in JAMA Netw Open. 2020 Apr 1;3(4):e206536]. JAMA Netw Open. 2020;3(2):e1921326. Published 2020 Feb 5. doi:10.1001/jamanetworkopen.2019.21326
[13] Kinnunen EM, Juvonen T, Airaksinen KE, et al. Clinical significance and determinants of the universal definition of perioperative bleeding classification in patients undergoing coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2014;148(4):1640-1646.e2. doi:10.1016/j.jtcvs.2014.07.040
[14] Held C, Asenblad N, Bassand JP, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes undergoing coronary artery bypass surgery: results from the PLATO (Platelet Inhibition and Patient Outcomes) trial. J Am Coll Cardiol. 2011;57(6):672-684.
[15] Zbrozek A, Magee G. Cost of Bleeding in Trauma and Complex Cardiac Surgery. Clin Ther. 2015;37(9):1966-1974. doi:10.1016/j.clinthera.2015.06.007
[16] Corral M, Ferko N, Hollmann S, Broder MS, Chang E. Health and economic outcomes associated with uncontrolled surgical bleeding: a retrospective analysis of the Premier Perspectives Database. Clinicoecon Outcomes Res. 2015;7:409-421. Published 2015 Jul 22. doi:10.2147/CEOR.S86369
[17] Hassan K, Kannmacher J, Wohlmuth P, Budde U, Schmoeckel M, Geidel S. Cytosorb Adsorption During Emergency Cardiac Operations in Patients at High Risk of Bleeding. Ann Thorac Surg. 2019;108(1):45-51. doi:10.1016/j.athoracsur.2018.12.032
[18] Development Update: U.S. FDA Grants Breakthrough Designation to CytoSorb for Removal of Ticagrelor During Cardiopulmonary Bypass in Emergent and Urgent Cardiothoracic Surgery. PRNewswire. April 20, 2020. Accessed October 17, 2021. https://www.prnewswire.com/news-releases/development-update-us-fda-grants-breakthrough-designation-to-cytosorb-for-removal-of-ticagrelor-during-cardiopulmonary-bypass-in-emergent-and-urgent-cardiothoracic-surgery-301043326.html
[19] ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29 – . Identifier NCT04976530, Safe and Timely Antithrombotic Removal – Ticagrelor (STAR-T) (STAR-T); 2021 July 26 [cited 2021 Oct 27]; Available from: https://clinicaltrials.gov/ct2/show/NCT04976530
[20] Cytosorbents Coporation Form 10-Q for Quarterly Period Ended June 30, 2021. Securities and Exchange Commission. Commission file number: 001-36792
[21] Russo JJ, James TE, Ruel M, et al. Ischemic and bleeding outcomes after coronary artery bypass grafting among patients initially treated with a P2Y12 receptor antagonist for acute coronary syndromes: Insights on timing of discontinuation of ticagrelor and clopidogrel prior to surgery. Eur Heart J Acute Cardiovasc Care. 2019;8(6):543-553. doi:10.1177/2048872617740832
[22] Second U.S. FDA Breakthrough Device Designation Granted to CytoSorbents’ DrugSorb-ATR™ Antithrombotic Removal System Adding the Removal of Market-Leading Direct Oral Anticoagulants During Urgent Cardiothoracic Surgery. PRNewswire. August 12, 2021. Accessed. October 27, 2021. https://www.prnewswire.com/news-releases/second-us-fda-breakthrough-device-designation-granted-to-cytosorbents-drugsorb-atr-antithrombotic-removal-system-adding-the-removal-of-market-leading-direct-oral-anticoagulants-during-urgent-cardiothoracic-surgery-301354007.html
[23] ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 Feb 29 – . Identifier NCT05093504, Safe and Timely Antithrombotic Removal – Direct Oral Anticoagulants Apixaban & Rivaroxaban (STAR-D) (STAR-D) ; 2021 Oct 26 [cited 2021 Oct 27]; Available from: https://clinicaltrials.gov/ct2/show/NCT05093504
[24] Guy GP, Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. MMWR Morb Mortal Wkly Rep. 2015;64(21):591-596.
[25] Guy GP, Machlin S, Ekwueme DU, Yabroff KR. Prevalence and costs of skin cancer treatment in the US, 2002–2006 and 2007–2011. Am J Prev Med. 2015;48:183–7.
[26] Stern RS. Prevalence of a history of skin cancer in 2007: results of an incidence-based model. Arch Dermatol. 2010 Mar;146(3):279-82.
[27] American Cancer Society. Cancer Facts & Figures 2021. Atlanta: American Cancer Society; 2021.
[28] Siegel RL, Miller KD, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021; doi: 10.3322/caac.21654.
[29] Howlader N, Noone AM, Krapcho M, et al (eds). SEER Cancer Statistics Review, 1975-2016, National Cancer Institute, Bethesda, MD, https://seer.cancer.gov/csr/1975_2016/, based on November 2018 SEER data submission, posted to the SEER website, April 2019.
[30] Skaggs R, Coldiron B. Skin biopsy and skin cancer treatment use in the Medicare population, 1993 to 2016. J Am Acad Dermatol. 2021;84(1):53-59. doi:10.1016/j.jaad.2020.06.030
[31] The Veriskin Solution. Accessed October 10, 2021. https://www.veriskin.com/technology.html
[32] National Center for Health Statistics. Percentage of having a wellness visit in past 12 months for adults aged 18 and over, United States, 2019. National Health Interview Survey. Generated interactively: Oct 22, 2021 from https://wwwn.cdc.gov/NHISDataQueryTool/SHS_2019_ADULT3/index.html
[33] Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4(1):13-37. doi:10.2217/mmt-2016-0022
[34] Al Kibria GM, Nemirovsky A, Sharmeen A, Day B. Age-stratified prevalence, treatment status, and associated factors of hypertension among US adults following application of the 2017 ACC/AHA guideline. Hypertens Res. 2019;42(10):1631-1643. doi:10.1038/s41440-019-0275-x
[35] Centers for Disease Control and Prevention, National Center for Health Statistics. Underlying Cause of Death 2018-2019 on CDC WONDER Online Database, released in 2020. Data are from the Multiple Cause of Death Files, 2018-2019, as compiled from data provided by the 57 vital statistics jurisdictions through the Vital Statistics Cooperative Program. Accessed at http://wonder.cdc.gov/ucd-icd10-expanded.html on Oct 14, 2021
[36] Wang G, Grosse SD, Schooley MW. Conducting Research on the Economics of Hypertension to Improve Cardiovascular Health. Am J Prev Med. 2017;53(6 Suppl 2):S115-S117. doi:10.1016/j.amepre.2017.08.005
[37] Zhang D, Wang G, Joo H. A Systematic Review of Economic Evidence on Community Hypertension Interventions. Am J Prev Med. 2017;53(6S2):S121-S130. doi:10.1016/j.amepre.2017.05.008
[38] Patel KV, Li X, Kondamudi N, et al. Prevalence of Apparent Treatment-Resistant Hypertension in the United States According to the 2017 High Blood Pressure Guideline. Mayo Clin Proc. 2019;94(5):776-782. doi:10.1016/j.mayocp.2018.12.033
[39] Muntner P, Hardy ST, Fine LJ, et al. Trends in Blood Pressure Control Among US Adults With Hypertension, 1999-2000 to 2017-2018. JAMA. 2020;324(12):1190-1200. doi:10.1001/jama.2020.14545
[40] Azizi M, Schmieder RE, Mahfoud F, et al. Endovascular ultrasound renal denervation to treat hypertension (RADIANCE-HTN SOLO): a multicentre, international, single-blind, randomised, sham-controlled trial [published correction appears in Lancet. 2018 Sep 8;392(10150):820]. Lancet. 2018;391(10137):2335-2345. doi:10.1016/S0140-6736(18)31082-1
[41] Azizi M, Sanghvi K, Saxena M, et al. Ultrasound renal denervation for hypertension resistant to a triple medication pill (RADIANCE-HTN TRIO): a randomised, multicentre, single-blind, sham-controlled trial. Lancet. 2021;397(10293):2476-2486. doi:10.1016/S0140-6736(21)00788-1
[42] Clinical Results, ReCor Medical. Accessed October 17, 2021. https://www.recormedical.com/clinical-results/
[43] Ettehad D, Emdin CA, Kiran A, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet. 2016; 387(10022):957-67
[44] National Cancer Institute SEER Program. Cancer stat facts: pancreatic cancer. https://seer.cancer.gov/statfacts/html/pancreas.html, Accessed October 14, 2021.
[45] Cronin KA, Lake AJ, Scott S, et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer. 2018;124(13):2785-2800. doi:10.1002/cncr.31551
[46] Doleh Y, Lal LS, Blauer-Petersen C, Antico G, Pishvaian M. Treatment patterns and outcomes in pancreatic cancer: Retrospective claims analysis. Cancer Med. 2020;9(10):3463-3476. doi:10.1002/cam4.3011
[47] Tramontano AC, Chen Y, Watson TR, et al. Pancreatic cancer treatment costs, including patient liability, by phase of care and treatment modality, 2000-2013. Medicine (Baltimore). 2019;98(49):e18082. doi:10.1097/MD.0000000000018082
[48] Bluestar Genomics Receives FDA Breakthrough Device Designation for First-of-Its-Kind Pancreatic Cancer Screening Test. Business Wire. March 31, 2021. Accessed Oct 28, 2021. https://www.businesswire.com/news/home/20210331005868/en/Bluestar-Genomics-Receives-FDA-Breakthrough-Device-Designation-for-First-of-Its-Kind-Pancreatic-Cancer-Screening-Test
[49] Could a Diabetes Diagnosis Help Detect Pancreatic Cancer Early? National Cancer Institute. Published July 7, 2021. Accessed October 14, 2021. https://www.cancer.gov/news-events/cancer-currents-blog/2021/pancreatic-cancer-diabetes-early-detection
[50] Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2020. Atlanta, GA: Centers for Disease Control and Prevention, U.S. Dept of Health and Human Services; 2020.
[51] Guler GD, Ning Y, Ku CJ, et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun. 2020;11(1):5270. Published 2020 Oct 19. doi:10.1038/s41467-020-18965-w
[52] Song CX, Yin S, Ma L, et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017;27(10):1231-1242. doi:10.1038/cr.2017.106
[53] Haan D, Bergamaschi A, et al. Genome-wide 5hmC profiles to enable cancer detection and tissue of origin classification in breast, colorectal, lung, ovarian, and pancreatic cancers. Journal of Clinical Oncology. 2021;39, no. 15_suppl (May 20, 2021) 3044-3044. doi: 10.1200/JCO.2021.39.15_suppl.3044
[54] CPT Category III Codes. American Medical Association. Accessed October 14, 2021. https://www.ama-assn.org/practice-management/cpt/category-iii-codes
[55] Local Coverage Determinations. Centers for Medicare and Medicaid Services. Updated October 19, 2021. Accessed October 20, 2021. https://www.cms.gov/Medicare/Coverage/DeterminationProcess/LCDs
[56] Tunis SR, Berenson RA, et al. Improving the quality and efficiency of Medicare program through coverage policy. Urban Institute. Published September 7, 2011. https://www.urban.org/research/publication/improving-quality-and-efficiency-medicare-program-through-coverage-policy
[57] Centers for Medicare & Medicaid Services (CMS). Decision Memo for Lumbar Artificial Disc Replacement (LADR) (CAG-00292R). [homepage on the internet]; 2007 Aug 14 Available from: https://www.cms.gov/medicare-coverage-database/view/ncacal-decision-memo.aspx?proposed=N&NCAId=197&NcaName=Lumbar+Artificial+Disc+Replacement+.
[58] McClellan M, Khan BB, Ray R, Lopez MH. RE: Medicare Coverage of Innovative Technology (MCIT) and Definition of “reasonable and Necessary” (CMS-3372-P), November 1, 2020. https://www.3d-dartmouthsymposium.org/wp-content/uploads/2020/11/Duke-Margolis-Comment_CMS-3372-P.pdf