Tags: Perspective
Cheyenne Ariana Erika Modina, Sandra Waugh Ruggles, and Josh Makower, Stanford University
Contact: emodina@stanford.edu>
Abstract
What is the message?
The study highlights challenges in the regulatory pathway for cell and gene therapies from the perspective of investors and innovators. These include FDA staff turnover, a lack of transparency in the approval process, and administrative inefficiencies. Addressing these regulatory challenges, improving communication, and enhancing early engagement between the FDA and stakeholders will accelerate the development and approval of this class of life-saving therapies.
What is the evidence?
A quantitative survey across cell and gene therapy investors and innovators responsible for their companies’ investor profiles or product portfolios.
Timeline: Submitted: October 15, 2024; accepted after review November 11, 2024.
Cite as: Cheyenne Ariana Erika Modina, Sandra Waugh Ruggles, and Josh Makower. 2024. Regulatory Pathway for Cell and Gene Therapies in the United States: Perspectives from Innovators and Investors. Health Management, Policy and Innovation (www.HMPI.org). Volume 9, Issue 3.
Introduction
Cell and gene therapies (CGTs) show promise as curative treatments for various diseases that currently have few existing treatment options.1 In 2017, the Food and Drug Administration (FDA) approved the first gene therapy in the United States,2 and there are currently more than 30 FDA-approved cell and gene therapies.3 However, the field has faced many barriers in developing these therapies and ensuring their safety and effectiveness. Historically, a high percentage of Investigational New Drug (IND) applications for CGTs have been terminated after years of development. From 1989 to 1999, 96% of IND applications were terminated after an average duration of 8.6 years, and from 1999 to 2009, 67% were halted after 7.5 years.3
Recognizing the challenges and difficulties of this therapeutic space, the FDA has issued multiple guidance documents for product development, clinical trials, and long-term follow-up to streamline the regulatory review process. This effort reflects the FDA’s understanding that the high development costs and small patient populations of cell and gene therapies can disincentivize companies from pursuing life-saving treatments.4 They also focused on collaborating with manufacturers and supporting small sponsors, such as academic investigators, who may not have the scale to conduct a clinical trial independently. To further increase the availability of CGTs, the FDA has set a goal of approving 10-20 CGTs annually by 2025; yet, as of 2023, only five CGTs have been approved, highlighting the persistent challenges in the development and regulatory pathway.5 They acknowledged that additional organizational support is needed to meet the demands of the surge of CGTs in development.6
These barriers may delay the accessibility of treatments to patients and the impact of future investments and innovations in the field. With rapid advancements in CGTs, regulatory processes must balance this with safety and efficacy. The research aims to describe the perspectives of investors and innovators in the regulatory pathway of cell and gene therapies. It describes the typical duration from first contact with the FDA to approval and identifies affected disease states due to regulatory challenges.
Methodology
The research involved a descriptive landscape study that surveyed both investors and innovators or manufacturers.
A modified version of the Centers for Disease Control and Prevention (CDC)’s Policy Analytical Framework was used for this research.7 Tailored to “Domain 2: Policy Analysis” within the framework, survey questions focusing on economic impact, feasibility, and public health impact were formulated. These three pillars describe the possible impact of the policy on investors and innovators. Figure 1 refers to the modified theoretical framework.
Figure 1. Theoretical framework.7

Using the theoretical framework, survey questions for each criterion were categorized among investors and innovators or manufacturers (see Appendix 1). A literature review on the policy topic informed the survey development between the two groups. Key informant interviews were conducted to validate the survey questions. Qualtrics XM (Qualtrics Version April 2024, Provo, UT)[*]8 was used to structure the survey questions for dissemination.
The survey was sent out to the National Venture Capital Association (NVCA and Biotechnology Innovation Organization (BIO), specifically for those who invest or work on cell and gene therapies, where both organizations either sent the survey directly to their members or posted it on social media using a convenience sampling approach. This approach targeted a broad representation of large, medium, and small-scale manufacturers. The inclusion criteria are:
- Must be an investor or an innovator in cell and gene therapy in the last five years
- Must be responsible for the research and development (R&D) pipeline or selecting the company’s product portfolio if the respondent is an innovator
This research received a “Notice for Exempt Review” under Stanford University’s Panel on Human Subjects in Medical Research (eProtocol 73552) on January 24, 2024. The survey underwent pretesting on Qualtrics XM (Qualtrics Version April 2024, Provo, UT)[*]8 and was distributed via email. Weekly follow-ups were implemented to achieve the desired sample size from all stakeholder groups. This structured approach aimed to enhance response rates and data reliability. A total of 143 respondents completed the pre-screening survey, and 141 eligible participants answered the pre-screening questions. Ninety-six participants completed the survey, giving a click-through to completion rate of 68%. Each IP address was checked to avoid duplicates. Twenty-six participants passed the inclusion criteria. Appendix 2 outlines the number of respondents and dropouts throughout the survey.
Data from Qualtrics XM (Qualtrics Version April 2024, Provo, UT)[*]8 was extracted to Google Sheets for cleaning, coding, and anonymization. Descriptive statistics were generated for the quantitative survey questions.
Study Results
Characteristics of Respondents
A total of 143 respondents began the pre-screening survey, of which 141 eligible participants completed the pre-screening questions. Out of these, 122 were chosen based on inclusion criteria that required them to be healthcare investors or innovators. Among all participants, a substantial majority of 67% are either innovators or biotech manufacturers, while healthcare investors make up 21%. Respondents who identified as “neither” (12%) were excluded from the analysis.
Table 1. Total survey respondents Survey respondents and demographic characteristics

Among investors, 91% of respondents are private equity or venture capital investors, while 9% are angel investors or represent family offices. The majority (39%) of participants managed a fund of between $10 and$100 million, with a relatively even distribution across all fund sizes: less than $10 million (22%), $100-500 million (26%), and more than $500 million (13%). Most investments span from pre-seed to Series B stages. About 37% of investors have funded Series A companies, 31% in pre-seed, and 24% in Series B. Fewer than 10% of respondents invest in Series C companies, and none in public companies.
Over the past five years, more than half of the respondents (74%) have invested in companies developing cell and gene therapies (2018-2023). In contrast, from the respondent pool, only 30% of innovators were developing cell and gene therapies.
Among innovators, 86% of respondents belong to executive leadership, while 12% are from research and development. Others are from clinical affairs (1%) and reimbursement (1%). 52% are from very small companies (1-50 employees), 36% from small companies (50-500 employees), 8% from large companies (>10,000 employees), and 4% from midsize companies (500 to 10,000 employees). Their primary source of funding is from private equity or venture capital (45%), publicly traded companies (32%), grants (10%), others (5%), and angel investors (5%).
Investor and innovator respondents have indicated they have invested and manufactured most heavily in oncology (30%). In contrast, endocrinology, pulmonary diseases, and ophthalmology have the least investments. Meanwhile, no manufacturers are developing cell and gene therapies for endocrinology. See Appendix 3 for cell and gene therapy investments and R&D for cell and gene therapies by disease states.
Perspectives of Investors and Innovators on CGT Regulatory Timelines
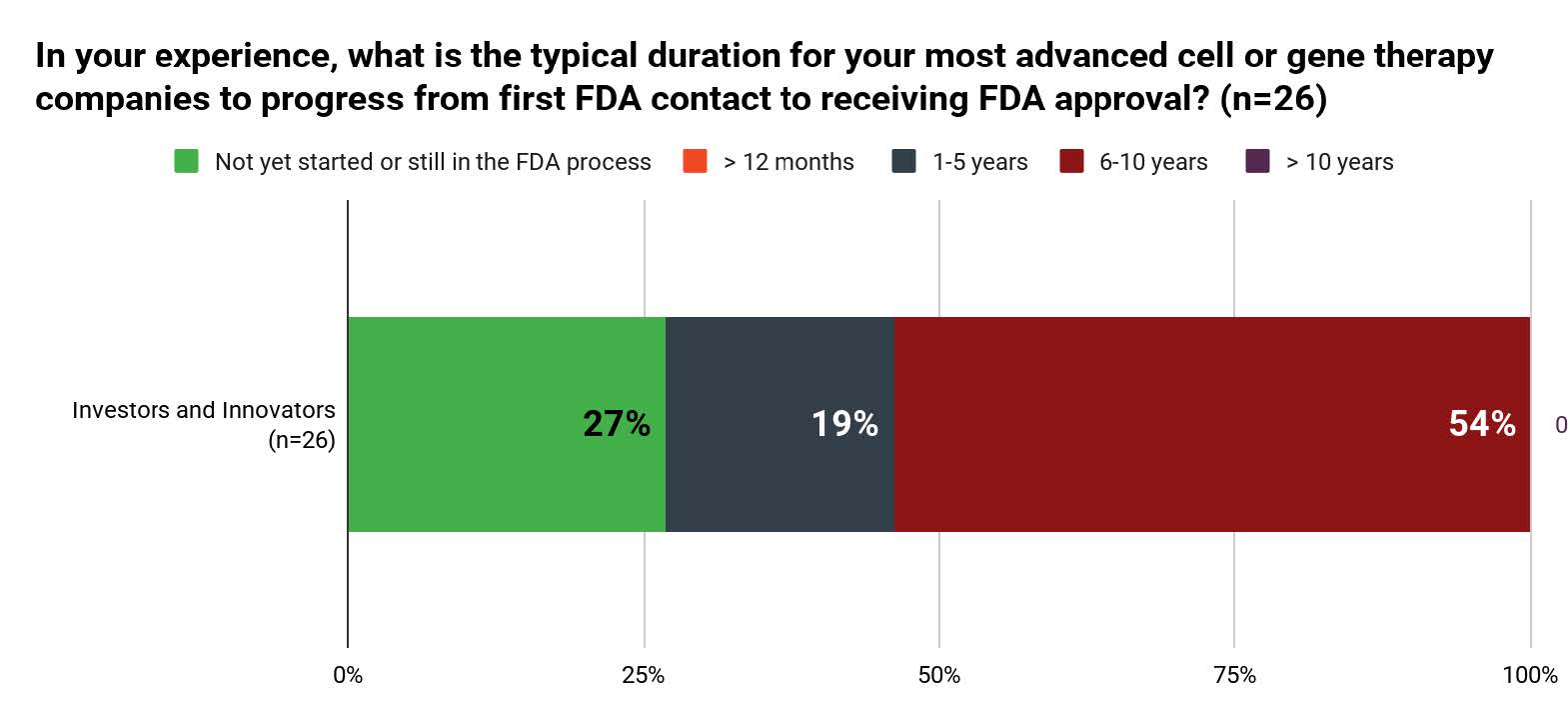
When asked, “What is the typical duration for your most advanced cell or gene therapy companies to progress from FDA contact to receiving FDA approval?” more than half (54%) of investors and innovators answered 6-10 years. Only 19% of the respondents answered 1-5 years, while 27% of the respondents have not started or are still in the FDA process. No respondent answered for more than ten years.
Figure 2. Typical Duration from First Contact to Approval Based on Investors and Innovators (n=26)
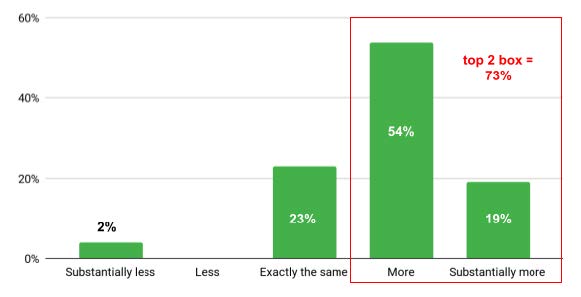
For roughly 73% of respondents, this duration was “more” and “substantially more” than the projected timeline, while 23% answered that it was “exactly the same” from what they projected. One respondent (7%) had gone through the approval process substantially less than the allocated time.
Figure 3. Allocated Timeline Projected (n=26)
When asked to rank the top three factors driving the regulatory timeline, 50% of investor and innovator respondents (n=26) mentioned that both “reviewer or key staff turnover” and “lack of transparency of the approval process” are the main reasons. Around 42% of respondents identified “administrative delays” as another factor that affects the regulatory timeline. Lastly, 38% of the respondents mentioned “changing of parameters for approval introduced during the review process.”
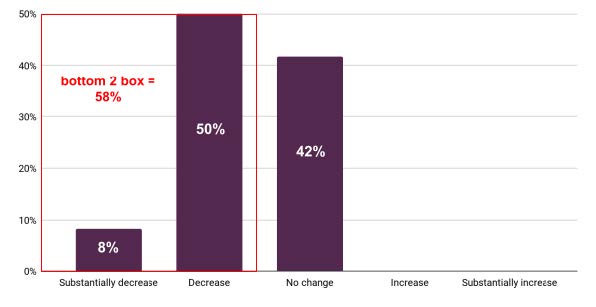
About 58% of investors indicated that these factors would “substantially decrease” and “decrease” their investments, while 42% indicated that this would not change their investments in CGTs. See Appendix 4 for complete survey results.
Figure 4. Change in Investments due to Policy (n=12)
Discussion
Despite the successes in the advancement of cell and gene therapies, attrition rates remain high.3 Our study and another study describe regulatory approval time for CGTs as typically achieved within 6 to 10 or 12 years.3 This timeline does not include pre-clinical research and development time, which reflects an even longer timeline for development. Our findings suggest that the time to FDA approval varies based on factors associated with the regulatory process itself rather than factors, such as pre-clinical development and clinical study design and execution, that could be predicted by the manufacturer. This highlighted that innovators did not account sufficiently for FDA timelines despite expecting a years-long process.
The primary challenge to achieving an efficient regulatory pathway, from the perspectives of investors and innovators, is reviewer or key staff turnover. There is a difference, however, in how turnover is measured between the two groups. The FDA defines “loss” as external to CBER. Thus a reviewer may be promoted or moved laterally within CBER and lost as a reviewer without being considered a gain or a loss by the FDA. CBER is working to develop analyses for accurate assessments of employee retention and engagement and has acknowledged and long struggled with the need for adequate staffing to achieve its goals of accelerating CGT approvals.9 They also acknowledge other retention challenges, such as career growth limitations, an overburdened workforce, and an increasing attrition rate, especially for employees nearing retirement.10 Staff are also highly sought after in the private industry, particularly in the fields of gene therapy and chemistry, which compounds the impact of retention challenges..4 Current retention efforts include leadership and development programs, such as FDA University, FDA Leadership Development Program, FDA Mentoring Program, DataForward, Project UpTech, the Environmental and Occupational Safety and Health (EOSH) Training Program10, and a student loan repayment program.11 Overall, the outlook is moving in a positive direction, with CBER announcing in June that it has accomplished 54% of its hiring goals for 2024.11,12
Other factors that manufacturers and investors believe lengthen regulatory approval processes include a lack of transparency in the approval process, administrative delays, theoretical safety concerns unsupported by data, and changing parameters for approval introduced during the review process. Therapies directly at cancer indications may be particularly impacted by changing parameters and administrative delays due to the long time needed for clinical trial participant recruitment. Clinical trials for cancer therapies average five years as compared to 3.5 years for rare genetic disorders and four months for infectious diseases.13 Despite these concerns, cancer indications are the most common investments (see Appendix 3) and were the most common disease focus in gene therapy clinical trials conducted from 2010 to 2020.13 CBER and the Center for Drug Evaluation and Research (CDER) started the Support for clinical Trials Advancing Rare Disease Therapeutics (START) Pilot Program to accelerate the development of CGTs that address an unmet clinical need resulting in disability or death within the first decade of life. Select program participants are working closely with the FDA for specific development issues like clinical study design, choosing patient populations and control groups, characterizing products, and using nonclinical information.14
Recommendations
Regulatory approval processes must ensure safety and efficacy while keeping pace with the rate of innovation as cell and gene therapies advance. This can be accomplished by strengthening the existing FDA pilot program, offering continuous development opportunities for reviewers, and enhancing the transparency of the regulatory process.
The FDA has made significant progress with the START Pilot Program, advancing the development and regulatory approval of CGTs through technical assistance and enhanced interactions with manufacturers. This program should facilitate more efficient development and help generate high-quality, compelling data to support a future marketing application.15 If successful, the FDA could expand this program to cell and gene therapies that address other disease states with long participant recruitment times. This could also expand outside of the CGT space for technologies with complex clinical endpoints and no available treatments. However, the FDA would need to dedicate additional teams and reviewers to support the program’s expansion and facilitate more efficient approvals.
The FDA may enhance reviewer expertise in cell and gene therapy by providing specialized training opportunities to ensure staffing stability. A clearer career development pathway for FDA reviewers with cell and gene therapy expertise could be established to encourage them to build long-term careers within CBER. CBER has identified strategies to retain staff and promote their development in its strategic plan,11 but an additional option may be to implement these initiatives with clear academic or leadership development opportunities for reviewers. This can be achieved by explicitly allocating time for these activities or exploring joint appointments with other institutions with CGT expertise. It is essential that management prioritize and protect the time allocated for career development for FDA reviewers. Reviewers should be able to engage in these activities without perceiving them as an additional burden but rather as a necessary component of professional growth and expertise development.
Improved communication and streamlining of administrative processes between the FDA and manufacturers are also essential for efficient regulatory processes. Early engagement meetings, such as INTERACT (Initial Targeted Engagement for Regulatory Advice on CBER/CDER ProducTs) meetings provide crucial guidance before definitive safety studies commence.16 In addition, the FDA could also update how staff transitions are managed and communicated. For instance, staffing metrics could include staff transitions to accurately account for reviewer change on a product-by-product basis. The FDA could better inform manufacturers when a reviewer has been promoted, reassigned or has left the FDA, as well. The SOPs for changing reviewers are publicly available, but increasing the transparency of Standard Operating Procedures (SOPs) for offboarding and documentation of all decisions could increase perceptions of transparency and consistency among manufacturers.
Future research on this topic could focus on a comprehensive evaluation of the specific causes of delays in the CGT regulatory approval process. These studies could involve qualitative interviews with stakeholders from biotechnology companies, investors, and the FDA. Additionally, a policy review of FDA processes may help improve communication and transparency in the regulation of cell and gene therapies. More targeted research could aim to streamline the review process by reducing redundant steps, improving coordination among reviewers, and incorporating more flexible timelines. This can be achieved by collaborating with companies to gather their feedback and co-develop a process that enhances transparency involving each stakeholder group.
Study Strengths and Limitations
Study strengths include the perspectives of both investors and manufacturers in the cell and gene therapy field, offering valuable insights into investment decisions and product pipeline development. As key decision-makers within their companies, they are well-qualified to answer the survey questions based on their current experience.
However, the study has limitations due to its small sample size, which could lead to response bias. The survey was distributed exclusively through the channels of the partner organizations, attracting participants who were more inclined to engage with the survey. Although an inclusion-exclusion criterion was applied, this does not entirely eliminate the possibility of response bias.
The use of convenience sampling may have excluded perspectives from other investors and innovators who were not reached through data collection. These experts might have contributed additional insights on the policy that were not captured in the survey results. Additionally, there were no open-ended questions to provide more comprehensive perspectives from innovators and investors.
Beyond regulatory approvals, other recommendations include improved coordination between the FDA and the Centers for Medicare and Medicaid Services (CMS) to better align the evidence required for both regulatory approval and reimbursement with manufacturers’ future needs. This is yet to be explored through a different research scope but can increase patient access.
Conclusion
The survey highlighted different factors that drive the regulatory timeline of cell and gene therapies. Despite advancements in the space, investors and innovators have indicated that prolonged regulatory timelines substantially impact investments and research and development for specific disease states. These findings underscore the challenges faced by stakeholders and highlight areas for improvement and opportunities for the continuous growth of cell and gene therapies in patient care.
Addressing key factors that drive regulatory timelines, such as staff turnover, lack of transparency, and regulatory inefficiencies, can achieve improvements to the regulatory pathway for CGTs. FDA is currently addressing challenges in the regulatory pathway through the START Pilot Program. In the future, the FDA could improve pathways for career development within CBER and strengthen expertise in cell and gene therapies to ensure knowledgeable and consistent personnel assigned to NDA review. Collaboration among investors, innovators, regulatory bodies, and patients is crucial to ensuring that life-saving technologies reach patients in a timely manner.
Notes
[*] The survey design and data collection for this paper was generated using Qualtrics software, Version January to April 2024 of Qualtrics. Copyright © 2024 Qualtrics. Qualtrics and all other Qualtrics product or service names are registered trademarks or trademarks of Qualtrics, Provo, UT, USA. https://www.qualtrics.com
References
- Stanford Medicine. Why Cell and Gene Therapy? Center for Definitive and Curative Medicine. Accessed April 24, 2024. https://med.stanford.edu/cdcm/CGT.html
- U.S. Food and Drug Administration. FDA approval brings first gene therapy to the United States. U.S. Food and Drug Administration. August 30, 2017. Accessed April 24, 2024. https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states
- Lapteva L, Purohit-Sheth T, Serabian M, Puri RK. Clinical Development of Gene Therapies: The First Three Decades and Counting. Mol Ther Methods Clin Dev. 2020;19:387-397. doi:10.1016/j.omtm.2020.10.004
- Bayer M. “Like salmon swimming upstream”: FDA’s Peter Marks lays out plan to boost gene therapy approvals. April 5, 2023. Accessed April 28, 2024. https://www.fiercebiotech.com/biotech/thats-failure-fdas-marks-says-gene-therapy-approvals-must-rapidly-increase
- Burr R. The FDA is at a crossroads on cell and gene therapies. STAT. November 20, 2023. Accessed April 24, 2024. https://www.statnews.com/2023/11/20/fda-cell-gene-therapies-biologics-evaluation-accelerated-approval/
- U.S. Food and Drug Administration. Statement from FDA Commissioner Scott Gottlieb, M.D. and Peter Marks, M.D., Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. U.S. Food and Drug Administration. March 24, 2020. Accessed April 24, 2024. https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics
- Center for Disease Control and Prevention. CDC’s Policy Analytical Framework | Policy, Performance, and Evaluation | CDC. May 9, 2023. Accessed October 30, 2023. https://www.cdc.gov/policy/polaris/policyprocess/policyanalysis/index.html
- Qualtrics. Qualtrics XM. Published online 2024. https://www.qualtrics.com
- Owermohle S. Official: FDA needs staff influx to meet gene therapy needs. STAT. December 4, 2023. Accessed October 2, 2024. https://www.statnews.com/2023/12/04/fda-gene-therapy-peter-marks/
- U.S. Food and Drug Administration. Report to Congress – Strategic Workforce Plan FYs 2023 to 2027. Published online 2023.
- U.S. Food and Drug Administration. PDUFA and BsUFA Quarterly Hiring Updates. Published online July 8, 2024. Accessed October 3, 2024. https://www.fda.gov/industry/prescription-drug-user-fee-amendments/pdufa-and-bsufa-quarterly-hiring-updates
- U.S. Food and Drug Administration. Center for Drug Evaluation and Research & Center for Biologics Evaluation and Research Net Hiring Data (FY 2023-2027). US Food Drug Administration. Published online July 11, 2024. Accessed October 3, 2024. https://www.fda.gov/industry/fda-user-fee-programs/center-drug-evaluation-and-research-center-biologics-evaluation-and-research-net-hiring-data-fy-2023
- Arabi F, Mansouri V, Ahmadbeigi N. Gene therapy clinical trials, where do we go? An overview. Biomedicine & Pharmacotherapy. 2022;153:113324. doi:10.1016/j.biopha.2022.113324
- U.S. Food and Drug Administration. Support for clinical Trials Advancing Rare disease Therapeutics (START) Pilot Program. US Food Drug Administration. Published online August 9, 2024. Accessed October 2, 2024. https://www.fda.gov/science-research/clinical-trials-and-human-subject-protection/support-clinical-trials-advancing-rare-disease-therapeutics-start-pilot-program
- U.S. Food and Drug Administration. FDA Launches Pilot Program to Help Further Accelerate Development of Rare Disease Therapies. U.S. Food and Drug Administration. August 9, 2024. Accessed October 15, 2024. https://www.fda.gov/news-events/press-announcements/fda-launches-pilot-program-help-further-accelerate-development-rare-disease-therapies
- Wills CA, Drago D, Pietrusko RG. Clinical holds for cell and gene therapy trials: Risks, impact, and lessons learned. Mol Ther Methods Clin Dev. 2023;31:101125. doi:10.1016/j.omtm.2023.101125
Appendix
Please download the Appendices here